Cancer
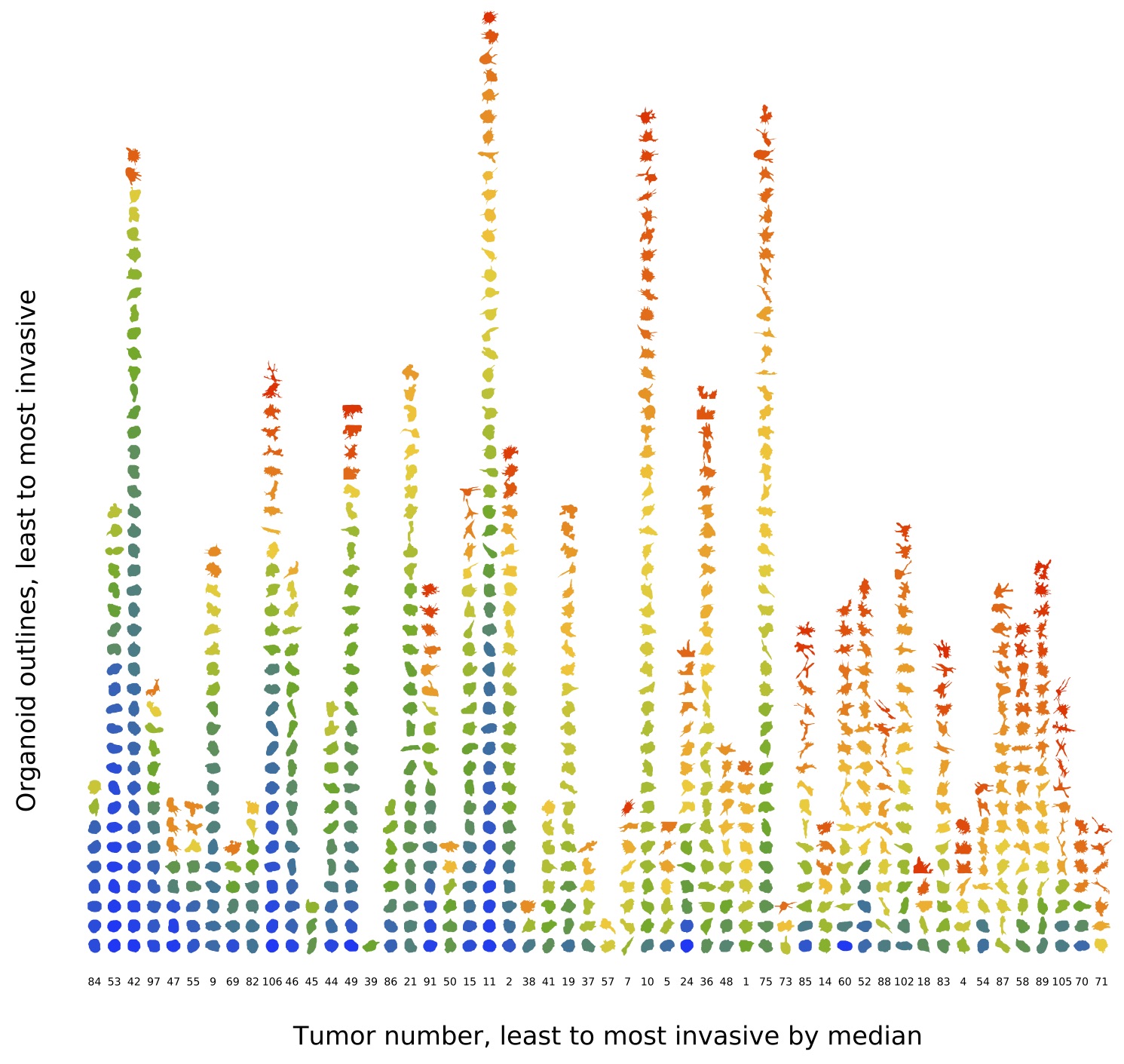
Joel Bader and Andy Ewald are Joint PI’s of the Johns Hopkins Cancer Target Discovery and Development (CTD2) Center, which was funded by the National Cancer Institute to identify new targets for metastatic breast cancer. Our study recruits women receiving breast cancer treatment at Johns Hopkins Hospital to donate biological specimens that will help reveal the molecular drivers of tumor invasion and metastasis. These phenotypes, rather than cell proliferation in the primary tumor, are the main cause of breast cancer mortality.
Our lab has developed image analysis methods that convert images to quantitative phenotypes for invasion. We are using these methods to identify genes that drive invasion in animal models and, with our ongoing study at Johns Hopkins Hospital, in people. We are applying network algorithms to trace the most likely pathways that connect upstream driver genes to downstream response genes with the goal of identifying new classes of therapeutic targets.
Synthetic biology
We develop methods for designing, building, testing, and analyzing DNA sequences that encode genes, pathways, chromosomes, and entire genomes. He is the computational leader of the international Saccharomyces cerevisiae 2.0 (Sc2.0) project, whose aim is to create a yeast cell with a synthetic genome. The milestone of synthesizing 6 of the 16 yeast chromosomes was highlighted in a series of 7 papers in the March 10, 2017, issue of the journal Science. Bader’s lab also develops technologies for biosafety and biosecurity, preventing escape of engineered life and identifying evidence of genetic engineering in genetic sequence data.

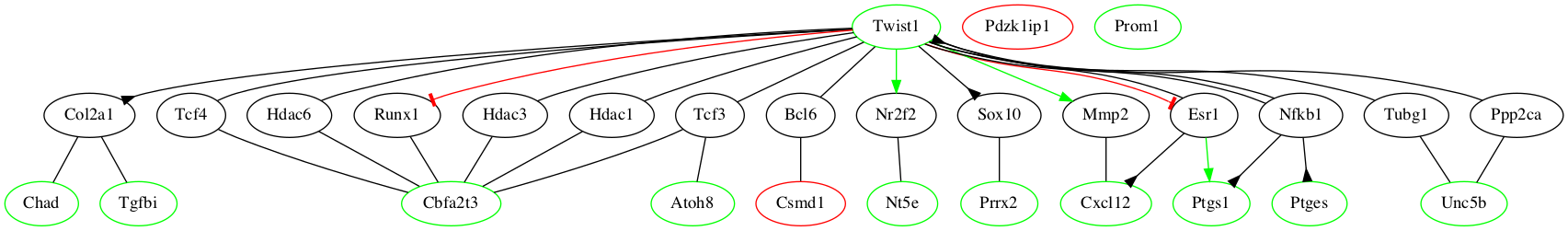
Algorithms for biological networks
The Human Genome Project defined many of the gene and protein components of our cells, but the connections between them and the rules for higher level organization into cells, tissues, organs, and organisms remain to be discovered. A grand challenge for systems biology is to determine what would be analogous to the wiring diagram for an electronic circuit: how do connections between genes and proteins lead to biological function? Our focus is on models that are interpretable and generalizable. These include generative models, which describe biological systems as observations drawn from a well-defined probability space, and physics-based models motivated by Hamiltonian systems in statistical mechanics.
We always build models because we want to answer real-world questions. These answers are unknown when we start our work, which means there is no ground truth for model assessment. In the absence of ground truth, statistical methods such as cross-validation are often used. This is a good idea, but it’s not the idea we rely on. Instead, essentially all of our work is done in the context of collaboration with wet-lab partners who want answers to the same questions. We work together using mathematical and computational methods to prioritize hypotheses, then perform experiments to accept or reject our proposals.
Genome-wide association studies
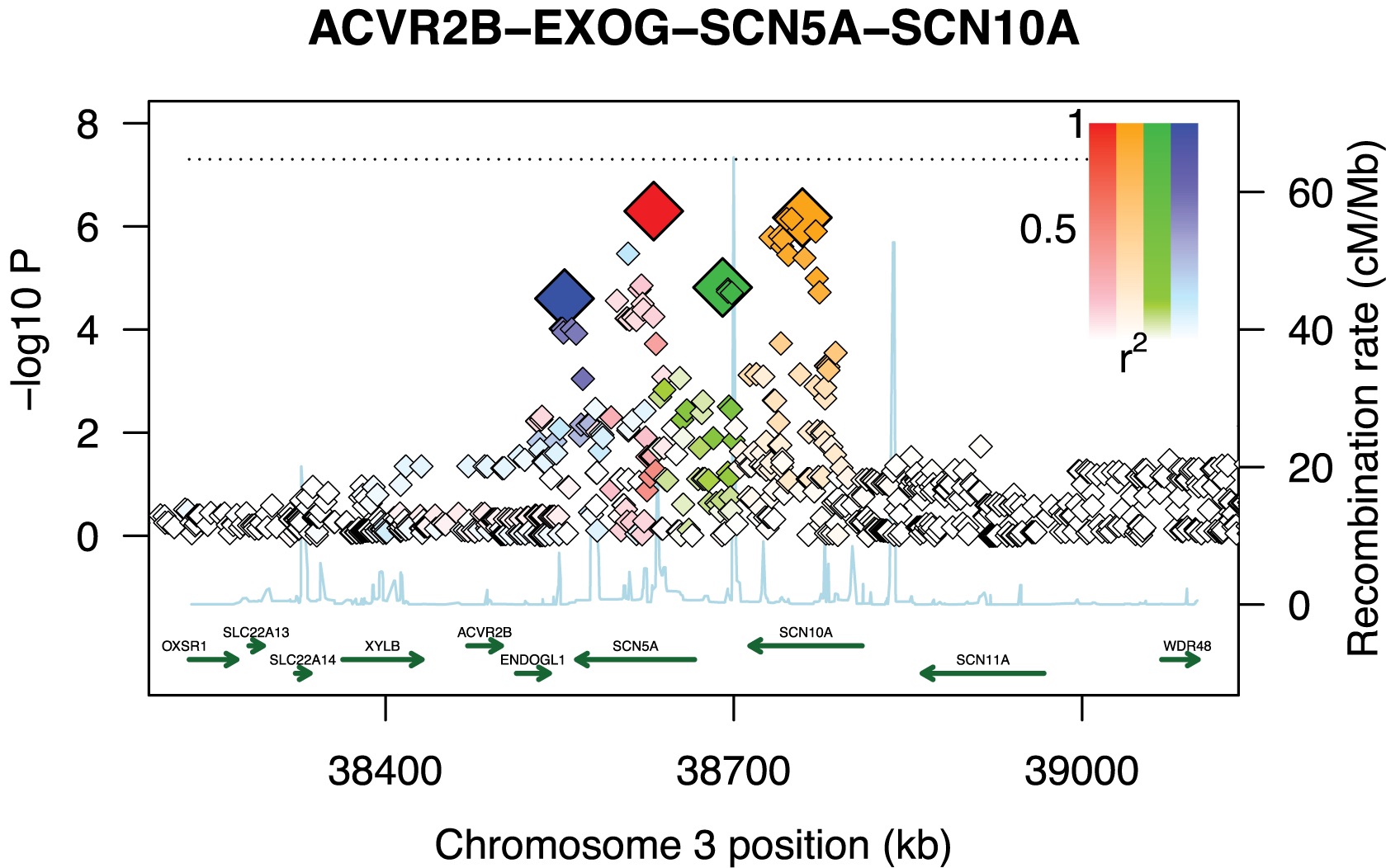
One of the main challenges in observational research is imputing causal order to a correlation or association. Just because A and B are correlated does not mean the A causes B, or even that B causes A. Maybe something upstream is responsible for both A and B, for example. This is important for human health because most studies of human disease are observational.
Genetic studies are special because we can usually assume that genetic variation is upstream of disease (the exception is when ethnic stratification or similar population structure creates a spurious association). And this is the reason for great interest in genome-wide association studies, which identify human genetic variants that affect disease risk.
We develop methods to improve the analysis of genome-wide association studies (GWAS). The methods we are most interested in are to move from the statistical realm of detecting an association to the biological realm of mechanism: what gene is responsible for the observed effect, what are its downstream targets, and could any of these have therapeutic potential.
Infectious disease
Many pathogens exhibit a phenotype known as persistence, the ability to persist in the presence of antibiotic treatment. This creates infections that are difficult to clear and can lead to large populations with latent disease. Long treatment times also lead to the possibility of selecting for pathogens with mutations that make them more resistant to treatment.
We work with the Karakousis lab at the Johns Hopkins School of Medicine to understand the persistence phenotype in Mycobacterium tuberculosis, and in other Mycobacterium species that are emerging pathogens. These studies include pathogen genetic screens to identify genetic requirements for persistence and joint analysis of host and pathogen biological networks.
